Today I've been exploring the Vibrio cholerae (the bacterial species that causes the disease cholera) genome data available in the Pathogenwatch website.
Finding out how many Vibrio cholerae genomes are in Pathogenwatch
I went to the Pathogenwatch website and clicked on 'Genomes' at the top. At the top, it says 'Viewing 73,294 of 73,294 genomes', which is all the genomes in Pathogenwatch for all species. To select V. cholerae genomes, I selected 'Vibrio' in the 'Genus' list on the left, and this then gave me a list of 390 V. cholerae genomes in a table:
There are several columns in the table:
Name: name of the assembly for the strain/isolate.
Organism: this is Vibrio cholerae in all cases.
Type: this is the group that this strain/isolate is classified into, using the MLST (multi-locus sequence typing) schema. The most common types are 69 (327 isolates/strains), followed by 737 (7 isolates/strains), 170 (5 isolates/strains), 48 (4 isolates/strains), 75 (3 isolates/strains), and so on.
Typing schema: this is MLST in all cases.
Country: this is the country that the strain/isolate was collected in (if available). The most isolates come from Mexico (92 isolates/strains), followed by China (59), Haiti (34), Nepal (25), India (20), Bangladesh (16) and Brazil (11), and so on. There are 57 strains/isolates with no country available, so we only have country information for 333 strains/isolates.
Date: this is the date the strain/isolate was collected (if available). These range from 1930 to 2011.
Access: this has values 'Public' or 'Reference'. I think the 'Reference' cases are reference genomes, and the rest are strains collected around the world.
The genomes listed as 'Reference' for V. cholerae are:
(1) Env-seawater, collected in 1982 in Brazil, with MLST type 79,
(2) Env-sewage, collected in 1978 in Brazil, with MLST type 48,
(3) 7PET_MiddleEastern, with MLST type 69,
(4) M66, with MLST type 71,
(5) W1_T1, with MLST type 69,
(6) W1_T2, with MLST type 69,
(7) W1_T3, with MLST type 69,
(8) W1_T4, with MLST type *b5a7,
(9) W1_T5, with MLST type 69,
(10) W1_T6, with MLST type 69,
(11) W1_T7, with MLST type 69,
(12) W1_T8, with MLST type 69,
(13) W1_T9, with MLST type 69,
(14) W1_T10, with MLST type 69,
(15) W1_T11, with MLST type 69,
(16) W1_T12, with MLST type 69,
(17) W1_T13, with MLST type 69.
I think that the MLST type *b5a7 for W1_T4 means that it didn't have a MLST type assigned, because the allele is not known for one of the loci.
Making a map for the sources of Vibrio cholerae genomes in Pathogenwatch
At the top of the list of 390 V. cholerae genomes, there are there three links, 'List', 'Map', 'Stats'. If you click on the 'Map', it gives you a map of where all the V. cholerae isolates/strains came from in the world:
You can see on the map that there are 92 isolates/strains from Mexico, 59 from China, 34 from Haiti, 25 from Nepal, 20 from India, 16 from Bangladesh, 11 from Brazil, and so on.
Getting assembly statistics for the Vibrio cholerae genomes in Pathogenwatch
If you click on the 'Stats' link at the top of the page (from the three links 'List', 'Map', 'Stats'), you will get assembly statistics for the 390 Vibrio cholerae genomes. There are several different assembly statistics available: genome length, N50, number of contigs, non-ACTG bases, and GC content.
If we look at the genome length, we see that the average genome size is 4,021,504.5 bases, about 4 Mbases:
I have labelled a couple of the assemblies that seem to have an unusually large or small assembly size. These might possibly be misassemblies, I think. In particular, the assembly SRR221551.contigs_spades seems to be huge (about 6.7 Mbases) compared to the rest.
If we look at the number of contigs, we see most assemblies have about 75 contigs (average 73.9), but that assembly SRR221551.contigs_spades also has a very large number of contigs, again suggesting the assembly is a bit dodgy:
Again, when we look at the GC content, we see that the assemblies have an average GC content of 47.5%, but assembly SRR221551.contigs_spades looks strange as it has an average GC content of about 53%:
Investigating the Haiti outbreak of Vibrio cholerae in Pathogenwatch
We can investigate the Haiti outbreak of Vibrio cholerae by creating a 'collection' of the V. cholerae isolates from Haiti in Pathogenwatch. I think that you need to log into Pathogenwatch using an email address to be allowed to do this. Then, in the 'map' view of all V. cholerae isolates from around the world, use the 'map selection tool' to draw a shape around Haiti, and this then selects the 34 V. cholerae isolates from Haiti:
In the list of assemblies that appears from Haiti (list on the left), select all the assemblies from Haiti, and then click on the 'Select genomes' button on the right and choose 'Create collection'. (Note: for some reason, I don't always see the 'Create collection' button, I'm not sure why.)
You can now see in the 'Collection view', that there is a map at the top showing Haiti, and a timeline at the bottom showing the dates for the isolates. In this case they are all for 2010. If you click on 'View tree', it will show a tree of the Haiti isolates also, which is a neighbour-joining tree based on the 'core' genes:
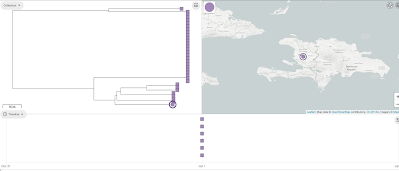
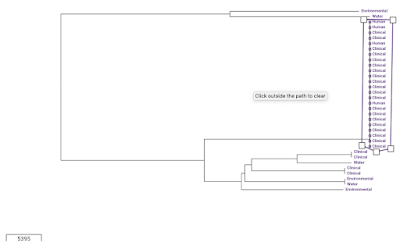
We can view the metadata for the assemblies in the collection by clicking on the 'Timeline' button at the bottom, and selecting 'Metadata' instead of 'Timeline'. One of the variables in the Metadata for the Haiti isolates is 'Source', which can take values such as 'Clinical', 'Environmental', and 'Water'. To show the 'Source' variable on the tree', we click on the 'Source' column in the Metadata table. Then click on the 'Settings' icon in the tree, and in the 'Nodes and labels' menu at the top left, we select 'Show leaf labels'. Note that the tree is a bit hard to read because the nodes are so big; to make them smaller click on the 'Nodes and labels' menu, and reduce the node size. We can see that there is one clade (which I've drawn a box around) of identical (or near identical) sequences that consists only of human/clinical isolates:
We can also view the MLST typing information for the isolates by selecting 'Typing' instead of 'Metadata'/'Timeline' in the menu on the bottom left. If we click on the 'Biotype' column, it shows in the tree that the highlighted clade all has the 'O1 pathogenic' biotype:
If we choose to display 'Reference' from the 'Typing' columns, we see that the isolates in this clade are closest to the W3_T12 reference, while the other isolates are closest to the 'Env_Sewage' reference:
And, if we choose the 'ST' column (MLST), we can see that the isolates in this clade are the 69 type, while the other isolates in the clade have a variety of MLST types:
Another interesting thing to look at is antibiotic resistance, and to do this we choose the 'Antibiotics' (instead of 'Timeline'/'Typing'/etc.). We should then see a table below with the resistance to different antiobiotics, with red dots indicating resistance:
As before, we can select a column, e.g. chloramphenicol resistance, and show which isolates are predicted to have chloramphenicol resistance on the tree, and we see that it is just the 'O1 pathogenic' clade that is predicted to have chloramphenicol resistance:











No comments:
Post a Comment